News
New Deep Learning Tool Accurately Classifies Tumor Types
Researchers from the NCI and Department of Energy (DOE) Collaboration have developed a deep learning tool for classifying human RNA-seq-based tumor types. This tool will be available for cancer researchers to use in the future, thanks to the Frederick National Laboratory for Cancer Research (FNLCR) Resource Curation Team and Argonne National Laboratory.
TULIP (Tumor cLassifIcation Predictor) is based on a one-dimensional, convolutional neural network framework derived from a previous NCI-DOE Collaboration resource, which aimed at accelerating advances in precision oncology and scientific computing.
Researchers updated the NCI-DOE resource, Tumor Classifier 1 (TC1), to include more tumor types (32 total) from NCI’s Genomic Data Commons. The team used CANcer Distributed Learning Environment (CANDLE), another NCI-DOE Collaboration resource, to optimize the underlying TC1 resource.
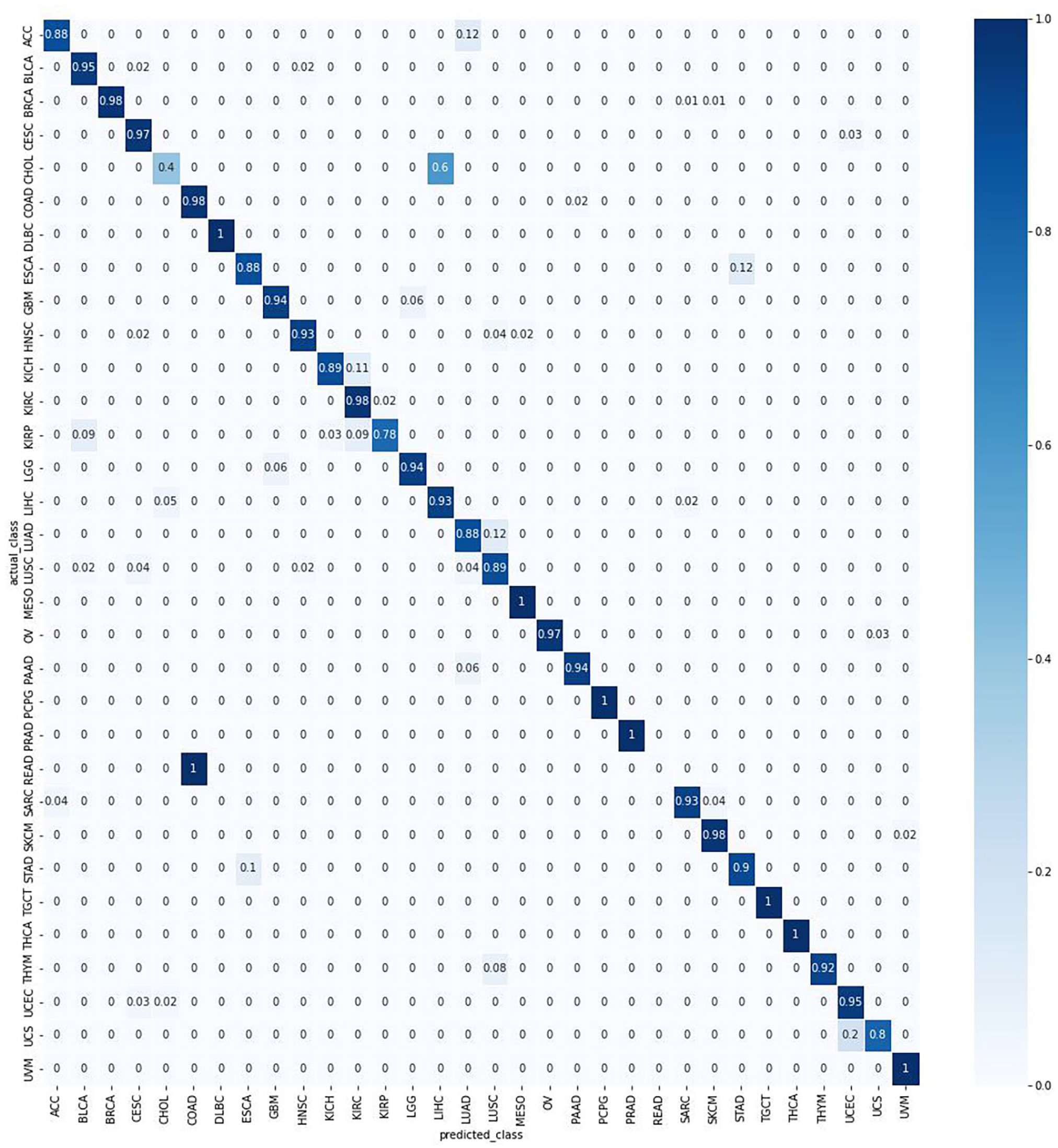
Overall, TULIP had at least a 95% accuracy on a test data set using protein-coding genes only. As shown in the confusion matrix in Figure 1, only two primary tumor types had below 50% accuracy. The reasons? Either the tumor types have highly similar RNA-seq profiles with other primary tumor types, or it’s due to a low sample size.
Dr. Satish Ranganathan Ganakammal, bioinformatics lead, explains that “the TULIP framework can be modified and adopted to perform machine learning (ML)-driven sample quality control in various RNA-seq analysis workflows.” The team recently adapted this framework to build an updated tumor tissue type classifier with canine-to-human common homologous genes. This was done to categorize canine RNA-seq samples based on the tumor tissue type. The work shows that developing cross-species ML models could have practical clinical applications, such as tumor subtype identification.
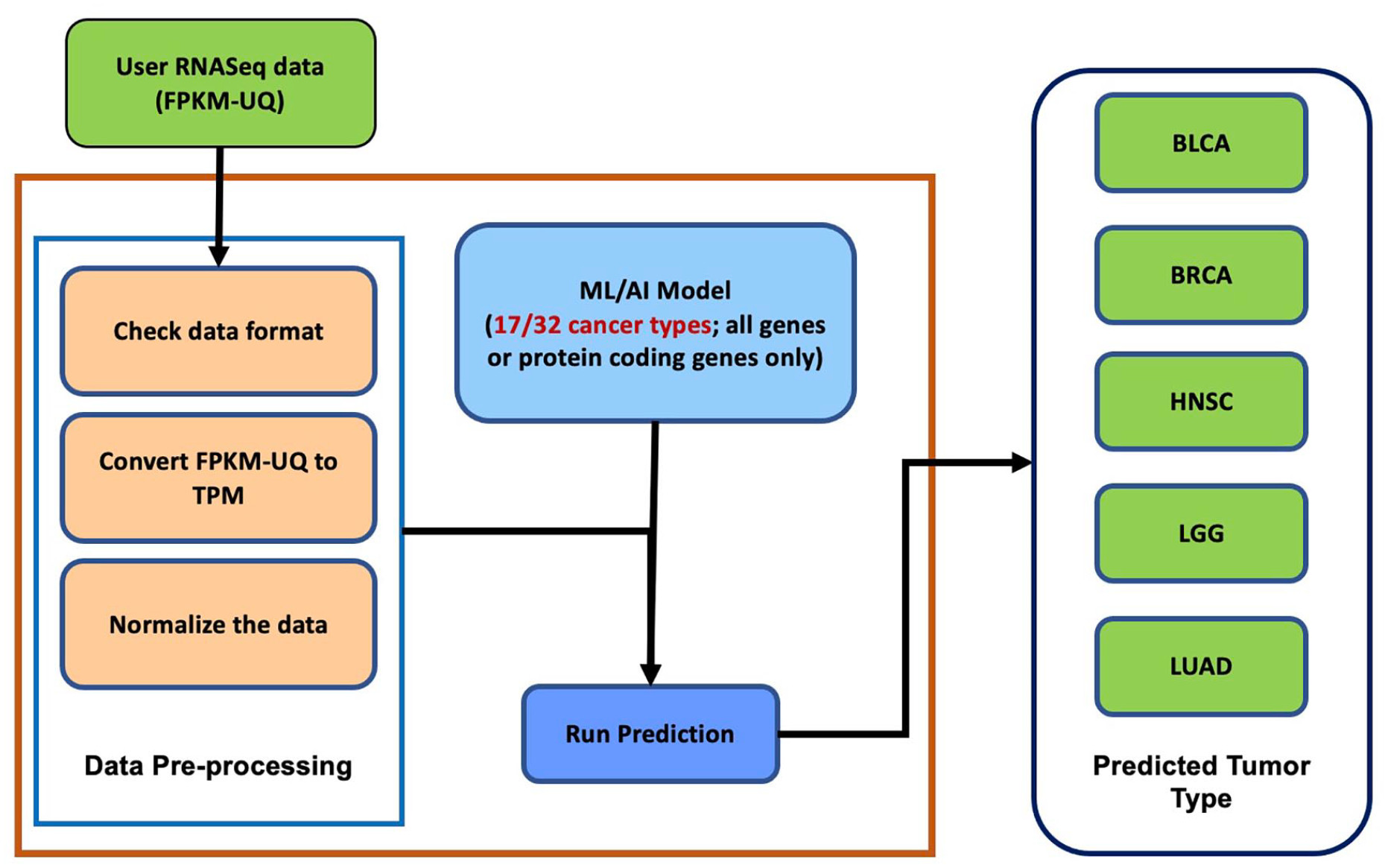
Matthew Beyers, FNLCR’s manager of the Resource Curation Team, shared, “As more data becomes available, we plan to expand our classification framework to include those new tumor tissue subtypes (see Figure 2). As a part of our functionality enhancement efforts, we plan to add normal tissue data to classify normal and tumor tissue type samples and include other data types such as variant data.”